检测到您当前使用浏览器版本过于老旧,会导致无法正常浏览网站;请您使用电脑里的其他浏览器如:360、QQ、搜狗浏览器的极速模式浏览,或者使用谷歌、火狐等浏览器。
 下载Firefox
下载Firefox
检测到您当前使用浏览器版本过于老旧,会导致无法正常浏览网站;请您使用电脑里的其他浏览器如:360、QQ、搜狗浏览器的极速模式浏览,或者使用谷歌、火狐等浏览器。
下载Firefox
线粒体是一种由双层膜包被的细胞器,负责提供细胞进行各种生命活动所需的能量。线粒体的蛋白质稳态维持对于其功能的正常行使至关重要。AAA+ ATPases(ATPases associated with a variety of cellular activities)在线粒体蛋白质质量控制中发挥着重要的作用。在线粒体内膜上存在两种AAA+ 蛋白酶复合物, m-AAA和i-AAA,它们分别朝向线粒体的基质(Matrix)和膜间隙(Intermembrane space,IMS),负责识别并降解线粒体复合物中错误组装和损坏的亚基,以及一些错误定位的膜蛋白。
线粒体还具有另外一种定位于IMS的AAA+ ATPase—CLPB (Perier et al., 1995)。CLPB的N端具有一个在AAA+家族成员很少见的ANK(Ankyrin repeat)结构域,其C端是一个典型的AAA+ ATPase模块。CLPB能够发挥去聚集酶(Disaggregase)的活性,在维持IMS蛋白质组稳态中发挥了重要作用 (Cupo and Shorter, 2020)。CLPB基因的突变与人类疾病密切相关,如3-甲基戊烯二酸疾病(3-MGA) (Capo-Chichi et al., 2015; Cupo and Shorter, 2020; Kanabus et al., 2015; Kiykim et al., 2016; Mroz et al., 2020; Saunders et al., 2015; Thevarajan et al., 2020; Wortmann et al., 2015)和重症先天性中性粒细胞减少症(SCN)(Warren et al., 2022)。在急性髓细胞性白血病(AML)中,CLPB蛋白的表达上调还介导了细胞对BCL-2抑制剂Venetoclax的耐药性 (Chen et al., 2019)。尽管CLPB对人类健康很重要,但现阶段对CLPB发挥功能的机制和结构的研究较少,其独特的ANK结构域的分子功能也并不清楚。
2023年2月6日,威尼斯wnsr666高宁课题组与合作者于PLOS Biology在线发表了题为Comprehensive structural characterization of the human AAA+ disaggregase CLPB in the apo- and substrate-bound states reveals a unique mode of action driven by oligomerization的研究论文。该研究利用冷冻电镜技术解析了人源CLPB在apo-和底物结合状态的结构,揭示了其在处理底物过程中的两种不同的寡聚状态。研究发现,CLPB的ANK结构域不仅负责维持CLPB独特高级组装形式,而且对去聚集酶的活性也不可或缺。该研究还通过质谱组学证明ANK直接参与多种线粒体底物的直接相互作用。这些结果揭示了CLPB作为线粒体中通用性的去聚集酶的独特生物物理性质,为以CLPB为靶点的各种线粒体相关疾病的药物研发提供了重要信息。
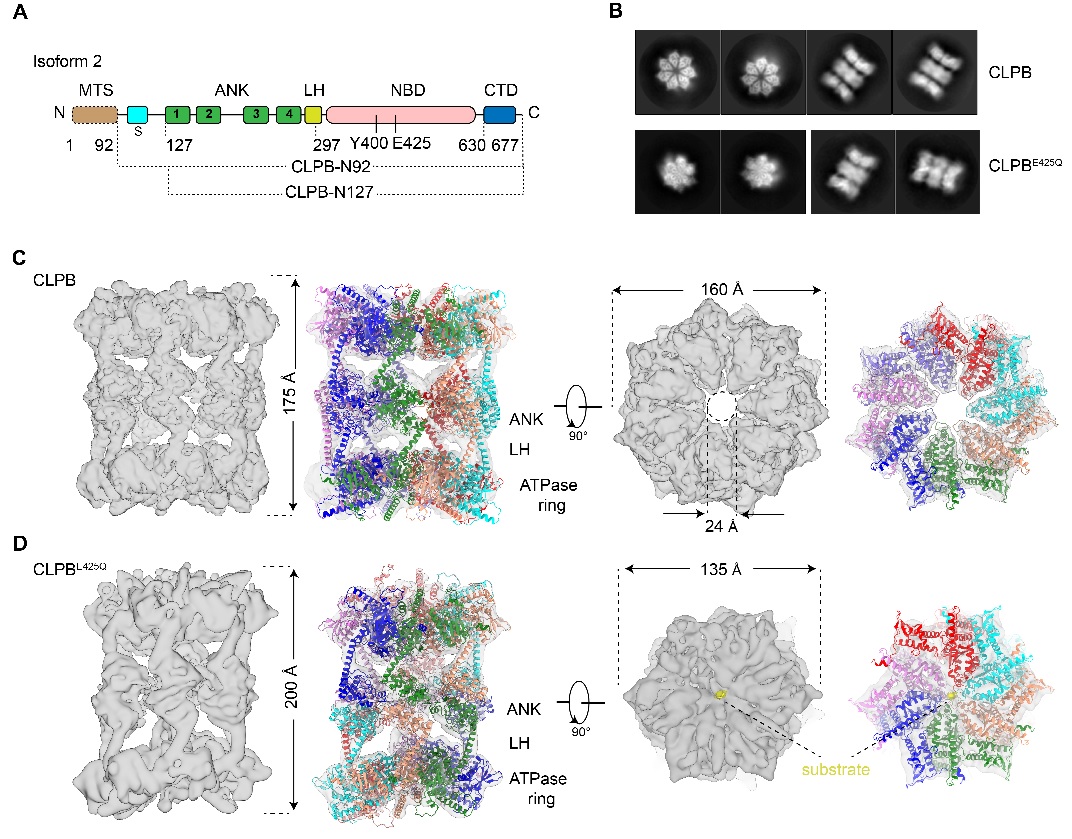
图1 apo-和底物结合状态的CLPB结构
首先,作者解析了apo状态CLPB的冷冻电镜结构。与经典的AAA+蛋白不同,apo状态的CLPB通过ANK结构域,以“头对头”的方式组装成同源十四聚体(双七聚体)。在apo状态中,中央孔道是空的,整个复合物呈假D7次对称。通过引入突变酶活性中心的突变(E425Q),作者解析了有内源底物结合的CLPB结构。惊奇的是,底物结合状态的CLPB形成了同源十二聚体(双六聚体)。与典型的AAA+ 蛋白质解聚酶或者去聚集酶类似,6个亚基呈螺旋阶梯状排列围绕着底物。AAA+蛋白家族中,七聚体的组织形式非常罕见。为了排除异源表达系统可能引起的假象,作者首先在哺乳动物细胞表达了野生型和突变的CLPB(WT和E425Q),并结合负染电镜分析其组装形式。研究发现,与E. coli表达的蛋白质的多聚形式完全一样:WT CLPB在不结合底物的情况下主要呈现双七聚体的形式;在体外加入模式底物Casein,可以诱导CLPB从双七聚体转变为双六聚体。这表明,双七聚体构象可能是CLPB的静息状态;在底物存在的情况下,底物和CLPB复合物之间的相互作用促进双六聚体的形成,而双六聚体具备更高的ATPase活性,是去聚集酶的活性状态。
由于双七/六聚体复合物分子间和分子内的高度动态性,结构的整体分辨率有限,无法分析底物的具体结合细节。为了探索CLPB结合底物的分子机理,论文解析了单独NBD的电镜结构以及ANK的晶体结构。结构表明CLPB保守的底物结合Loop与底物直接相互作用,围绕着底物呈螺旋阶梯状排列。基于ANK的高分辨结构,作者分析了介导双层结构的ANK结构域的界面,发现界面的某些残基的突变可以显著的降低CLPB的去聚集酶活性。最后,利用LC-MS/MS技术,作者对CLPB ANK结构域的线粒体互作蛋白组进行了定性和定量的分析,揭示了ANK结构域对底物的识别和选择。
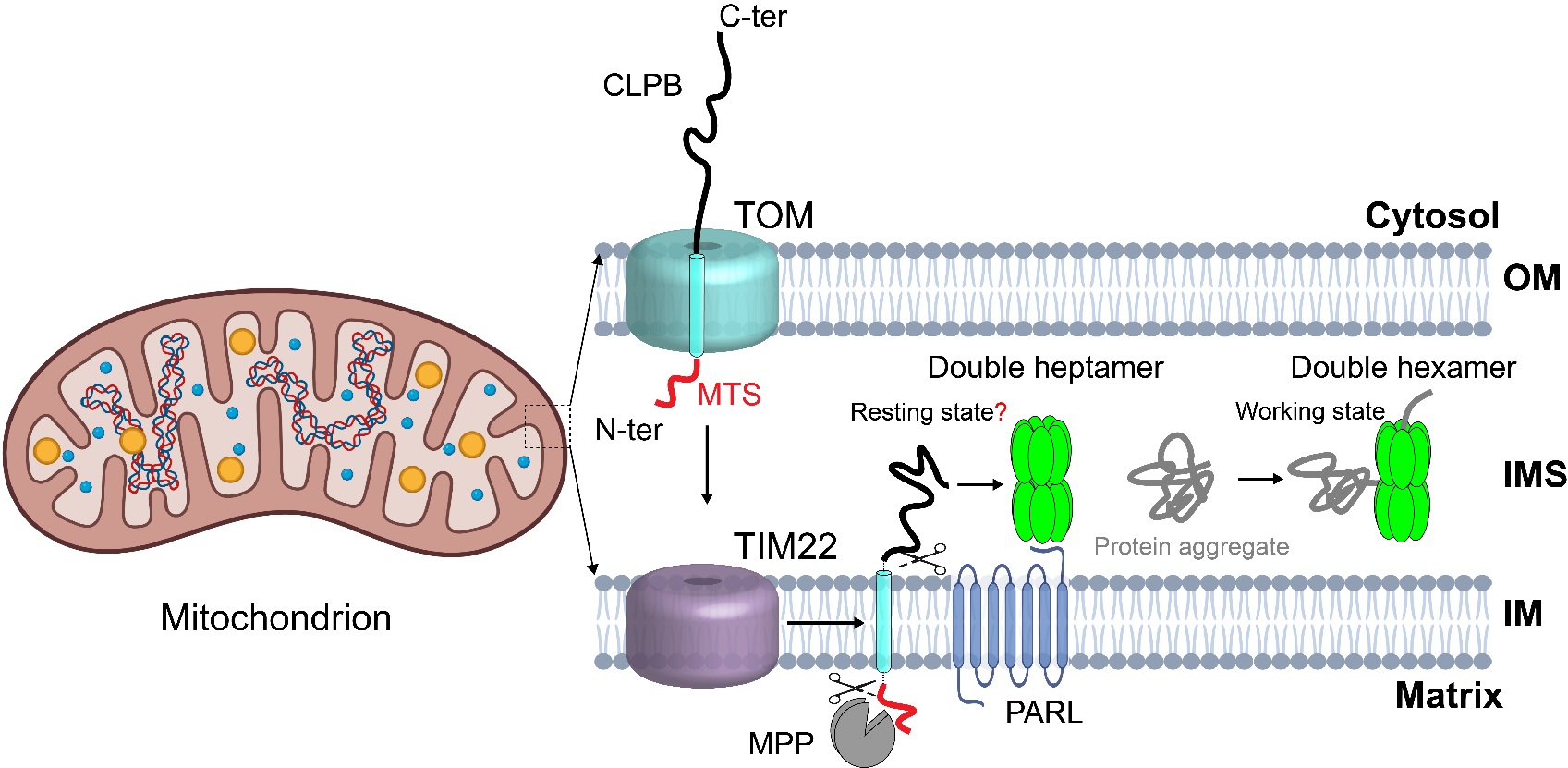
图2 CLPB的工作模型
综上,本项工作利用冷冻电镜结合生化、质谱等手段,对CLPB的结构和功能进行了系统性的研究,为理解CLPB在线粒体中发挥去聚集酶活性的分子机理提供了重要的理论基础,为后续设计CLPB的抑制剂来治疗相关疾病提供了重要的结构基础。
威尼斯wnsr666高宁教授、复旦大学威尼斯wnsr666林金钟教授为该论文的共同通讯作者,课题组吴大木博士研究生(2017级PTN)为本文的第一作者,刘燕博士(复旦大学)、戴余浩(2020级PTN)、王国鹏博士、逯国亮博士(复旦大学)对文章做出了重要贡献,陈燕博士、李宁宁博士在该工作中亦有贡献。该研究得到了国家自然科学基金、国家重点研发计划、启东-SLS创新基金、北大-清华生命科学联合中心、膜生物学国家重点实验室、昌平实验室的支持。威尼斯wnsr666冷冻电镜平台、电镜实验室、高性能计算平台、威尼斯wnsr666仪器中心及凤凰工程蛋白质质谱平台等多个仪器平台对本项目提供了重要的技术支撑。
原文链接:https://doi.org/10.1371/journal.pbio.3001987
参考文献:
Capo-Chichi, J.M., Boissel, S., Brustein, E., Pickles, S., Fallet-Bianco, C., Nassif, C., Patry, L., Dobrzeniecka, S., Liao, M., Labuda, D., et al. (2015). Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria. J Med Genet 52, 303-311.
Chen, X., Glytsou, C., Zhou, H., Narang, S., Reyna, D.E., Lopez, A., Sakellaropoulos, T., Gong, Y., Kloetgen, A., Yap, Y.S., et al. (2019). Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov 9, 890-909.
Cupo, R.R., and Shorter, J. (2020). Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. Elife 9.
Kanabus, M., Shahni, R., Saldanha, J.W., Murphy, E., Plagnol, V., Hoff, W.V., Heales, S., and Rahman, S. (2015). Bi-allelic CLPB mutations cause cataract, renal cysts, nephrocalcinosis and 3-methylglutaconic aciduria, a novel disorder of mitochondrial protein disaggregation. J Inherit Metab Dis 38, 211-219.
Kiykim, A., Garncarz, W., Karakoc-Aydiner, E., Ozen, A., Kiykim, E., Yesil, G., Boztug, K., and Baris, S. (2016). Novel CLPB mutation in a patient with 3-methylglutaconic aciduria causing severe neurological involvement and congenital neutropenia. Clin Immunol 165, 1-3.
Mroz, D., Wyszkowski, H., Szablewski, T., Zawieracz, K., Dutkiewicz, R., Bury, K., Wortmann, S.B., Wevers, R.A., and Zietkiewicz, S. (2020). CLPB (caseinolytic peptidase B homolog), the first mitochondrial protein refoldase associated with human disease. Biochim Biophys Acta Gen Subj 1864, 129512.
Perier, F., Radeke, C.M., Raab-Graham, K.F., and Vandenberg, C.A. (1995). Expression of a putative ATPase suppresses the growth defect of a yeast potassium transport mutant: identification of a mammalian member of the Clp/HSP104 family. Gene 152, 157-163.
Saunders, C., Smith, L., Wibrand, F., Ravn, K., Bross, P., Thiffault, I., Christensen, M., Atherton, A., Farrow, E., Miller, N., et al. (2015). CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria. Am J Hum Genet 96, 258-265.
Thevarajan, I., Zolkiewski, M., and Zolkiewska, A. (2020). Human CLPB forms ATP-dependent complexes in the mitochondrial intermembrane space. Int J Biochem Cell Biol 127, 105841.
Warren, J.T., Cupo, R.R., Wattanasirakul, P., Spencer, D.H., Locke, A.E., Makaryan, V., Bolyard, A.A., Kelley, M.L., Kingston, N.L., Shorter, J., et al. (2022). Heterozygous variants of CLPB are a cause of severe congenital neutropenia. Blood 139, 779-791.
Wortmann, S.B., Zietkiewicz, S., Kousi, M., Szklarczyk, R., Haack, T.B., Gersting, S.W., Muntau, A.C., Rakovic, A., Renkema, G.H., Rodenburg, R.J., et al. (2015). CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet 96, 245-257.
地址:北京市海淀区颐和园路5号
金光生命科学大楼
电话:010-62757794

北大生科官方微信

生声不息公众号